氮素代谢在植物的新陈代谢中占主导地位。植物组织中有机氮化物的含量随着植物的生理状况及环境条件的不同而发生变化。所以测定其含量,对研究植物的氮素吸收、运输和代谢规律,以及确定农产品的品质、营养价值等具有一定意义。
原理:
植物组织中的有机氮化物包括蛋白氮和非蛋白氮。非蛋白氮主要是氨基酸和酰胺,以及少量无机氮化物,是可溶于三氯乙酸溶液的小分子。可加入三氯乙酸,使其最终浓度为5%,将蛋白质沉淀出来,分别测定总氮、蛋白氮或非蛋白氮,通常只测定总氮或蛋白氨。
将植物材料与浓硫酸共热.硫酸分解为二氧化硫、水和原子态氧,并将有机物氧化分解成二氧化碳和水;而其中的氮转变成氨,并进一步生成硫酸铵。为了加速有机物质的分解,在消化时通常加人多种催化剂,如硫酸铜、硫酸钾和硒粉等。消化完成后,加人过量的Na0H,将NH4+转变成NH3,通过蒸馏把NH3导人过量的硼酸溶液中,再用标准盐酸滴定,直到硼酸溶液恢复原来的氢离子浓度。滴定消耗的标准盐酸摩尔数即为NH3的摩尔数,通过计算,可得出含氮量。以甘氨酸为例,上述反应如下:
蛋白质是一类复杂的含氮化合物,每种蛋白质都有其恒定的含氮量(约在14%-18%,平均约含氮16%),所以,可用蛋白氮的量乘以6.25( 100/16=6.25),算出蛋白质的含量。若以总氨含量乘以6.25,就是样品的粗蛋白含量。
试样中若含有硝态氮时,首先要使硝态氮还原为铵态氮,可加人水杨酸和硫代硫酸钠,使硝态氮与水杨酸在室温下作用生成硝基水杨酸.再用硫代硫酸钠(Na2S2O3)或Zn粉使硝基水杨酸转化为铵盐。由于水杨酸与硫代硫酸钠会消耗一部分硫酸,所以消化时的硫酸用量要酌情增加。
材料、仪器设备及试剂
(一)材料
各种干燥、粉碎、过筛(60-80目)的植物样品。
(二)仪器设备
消化管,微量凯氏定氮蒸馏装置一套(图1),三角烧瓶,微量滴定管,量筒,容量瓶,烧杯,移液管等。
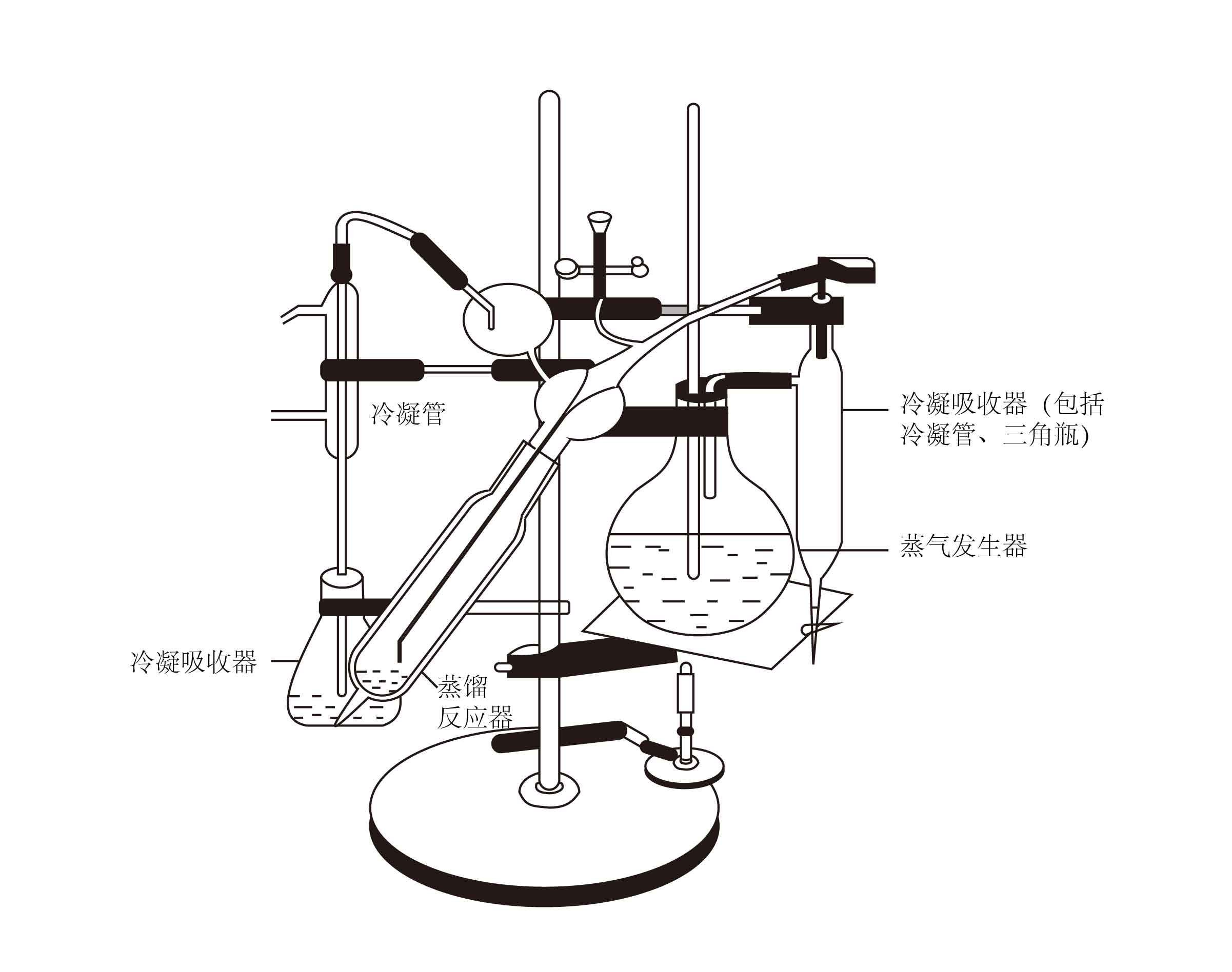
图1 微量凯氏定氮装置
(三)试剂
(1)浓硫酸(AR级)。 (2)5%三氯乙酸。 (3)30%过氯化氢。 (4) 40% NaOH。
(5)混合催化剂:K2SO4: CuSO4•5H2O=3:1或Se:CuSO4•5H2O: K2SO4=1:5:50,充分研细、混匀,贮于广口瓶中备用。实验步骤
(一)样品提取分离
准确称取烘至恒重的样品0.1000g-0.5000g (依样品含氮量而定,含氮1-3mg为宜),置10mL离心管中,加入5mL5%三氯乙酸,90℃水浴中浸提15min,不时搅拌,取出后用少量蒸馏水冲洗玻棒,待溶液冷却后,4000r/min离心15 min,上清液弃去。并用5%三氯乙酸洗沉淀2-3次,离心,弃去上清液,最后用蒸馏水将沉淀无损地洗人铺有滤纸的漏斗上,去掉滤液后,将沉淀和滤纸在50℃下烘干,用于蛋白氮的测定。
(二)样品的消化
取4支清化管编号。1号管直接放人称好的材料用于测定总氮,2号管放人上述烘千的滤纸和沉淀,用于蛋白氮的测定,3号管放人同样滤纸一张,4号管不加任何样品作为空白对照,注意将样品放人消化管底部。
向各消化管加浓硫酸5mL,混合催化剂0.3-0.5g,将样品浸泡数小时或放置过夜后,在管口盖一小漏斗,放在远红外消煮炉上加热消化。开始时温度可稍低,以防止内容物上升至管口。泡沫多时,可从小漏斗加人2-3滴无水乙醇。到管口出现白色雾状物时,泡沫已不再产生,此时可逐渐升温,使内容物达到微沸,直到消化液变为清澈透明为止。消化过程中.若在消化管.上部发现有黑色颗粒时,应小心地转动消化管,用消化液将它冲洗下来.以保证样品消化完全。消化过程约需2-3h。
(三)定容
消化完毕,待溶液冷却后,沿管壁仔细加入10 mL左右无氨蒸馏水,以冲洗管壁,再将消化液小心转人100 mL容量瓶中。以无氨水少量多次冲洗消化管,冼涤液并人容量瓶。冷却后用无氨水定容至刻度,混匀备用。
(四)蒸馏及滴定
蒸馏及滴定过程可分以下几步进行:
1.仪器的洗涤
先经一般洗涤后,还要用水蒸气洗涤。可按下列方法进行蒸气洗涤。先在蒸气发生器中加人2/3体积的蒸馏水(事先加入几滴浓硫酸,使其酸化,加入甲基红指示剂,并加人少许沸石或毛细玻璃管以防止爆沸)。打开漏斗下的夹子,用电炉或酒精炉加热至沸腾,使水蒸气通人仪器的各个部分,以达到清洗的目的。在冷凝管下端放置一个三角瓶接收冷凝水,然后关紧漏斗下的夹子,继续用蒸气洗涤5 min。 冲洗完毕,夹紧蒸气发生器与收集器之间的连接橡胶管,蒸馏瓶中的废液由于减压而倒吸进人收集器,打开收集器下端的活塞排除废液。如此清洗2-3次,再在冷凝管下端换放一个盛有硼酸-指示剂混合液的三角瓶,使冷凝管下口完全浸没在液面以下0.5cm处,燕馏1-2min,观察三角瓶内的溶液是否变色。如不变色,表示蒸馏装置内部已洗干净。移去三角瓶,再蒸馏1-2min,用蒸馏水冲洗冷凝管下口,关闭电炉,仪器即可供测定样品使用(图1)。
2.标准硫酸铵测定
为了熟悉蒸馏和滴定的操作技术,并检验实验的准确性,找出系统误差,常用已知浓度的标准硫酸铵测试三次。在三角瓶中加人20mL硼酸-指示剂混合液,将此三角瓶承接在冷凝管下端,并使冷凝管的出口浸人溶液中。注意在加样前务必打开收集器活塞,以免三角瓶内液体倒吸。准确吸取2mL硫酸铵标准溶液,加到漏斗中,小心打开漏斗下的夹子,使硫酸铵缓慢流人蒸馏瓶中,用少量蒸馏水洗涤漏斗三次(每次约1-2mL),一并放人蒸馏瓶中。然后用量筒向漏斗里加人10mL40%的氢氧化钠溶液,使碱液慢慢流人蒸馏瓶中,待碱液尚未流完时,将夹子夹紧,向漏斗中加约5mL蒸馏水,再慢慢打开夹子,使一半蒸馏水流入蒸溜瓶,另一半留在漏斗中作水封。关闭收集器活塞,加热蒸汽发生器,开始蒸馏。三角瓶中硼酸一指示剂混合液由于吸收了氨,由紫红色变成绿色。自变色时起,再蒸馏3-5min后,移动三角瓶,使瓶内液面离开冷凝管下口约1cm,并用少量蒸馏水洗涤冷凝管下口,继续蒸馏1min,移开三角瓶,盖上表面。
按上述方法再用标准硫酸铵测定两次。另取2ml蒸馏水代替硫酸铵溶液进行空白测定。将各次蒸馏的三角瓶一起滴定。取三次滴定的平均值进行含氮量的计算,并将结果与标准值(即加入硫酸铍中的含氮量)进行比较,得到氮的回收率。
每次蒸馏完毕,移去电炉,夹紧蒸气发生器与收集器之间的橡胶管,排除废液并用蒸馏水冲洗漏斗几次,废液排出,如此反复冲洗干净后,即可进行下一个样品的蒸馏。
3.样品及空白的蒸馏
准确吸取稀释后的消化液5mL,通过漏斗加入到蒸馏瓶中,再用少量蒸馏水洗涤漏斗3次,其余操作按标准硫酸铵的蒸馏进行。
4.滴定
样品与空白蒸馏完毕后,一起进行滴定。选用微量酸式滴定管,加人0.010 0mol/L标准盐酸溶液进行滴定,直至二角瓶中硼酸-指示剂混合液由绿色变回淡紫红色即为滴定终点,记录盐酸的用量。
结果计算

式中: 0.0100为标准盐酸的浓度; 100为消化液定容体积; 5为蒸馏时吸取消化液的体积;2.0为蒸馏取用标准硫酸铵的体积;0.6表示标推硫酸铵溶液含氮量为0.6mg/mL;V为标准硫酸铵的滴定值/mL;V,为空白的滴定值/mL;V为蛋白氮的滴定值/mL;V2为滤纸的滴定值/mL,;V,为总氮的滴定值/mL;W为样品重量/g。
注意事项
(1)在蒸馏过程中,切勿关闭电炉,否则会引起硼酸液的倒吸。
(2)环境中氨气的含量要低。
(3)定氮仪各连接处绝对不能漏气。
(4)所用橡皮塞、管用前均需处理。其方法是:浸在10%氢氧化钠溶液中煮沸约10min.再经水洗和水煮10min,最后冲洗数次。
独立公正 方法科学 规范严谨 服务周到